
{kind=link}
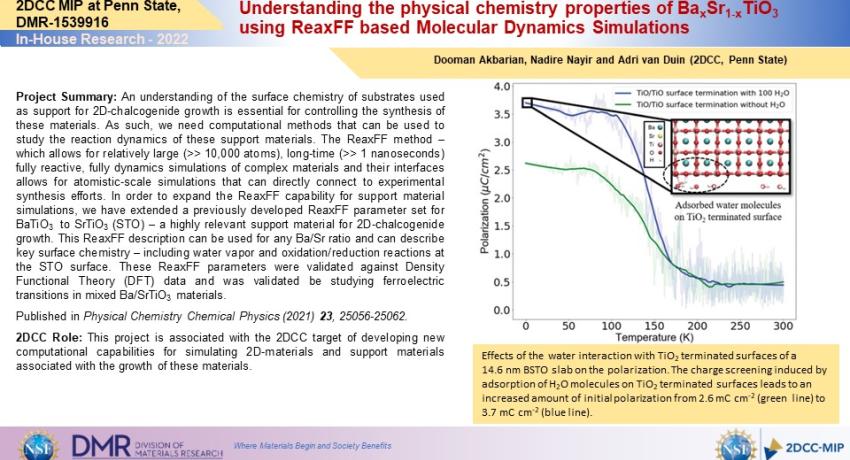
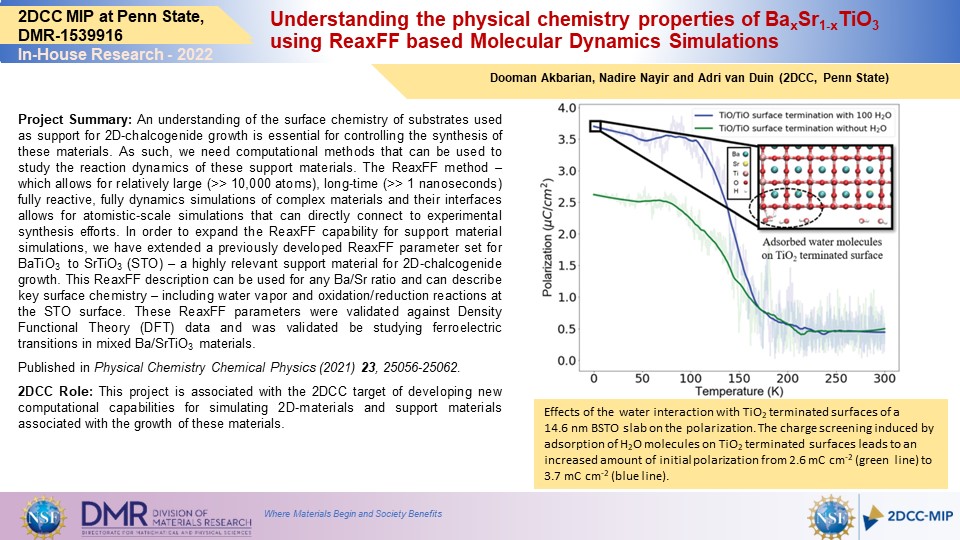
Project Summary: An understanding of the surface chemistry of substrates used as support for 2D-chalcogenide growth is essential for controlling the synthesis of these materials. As such, we need computational methods that can be used to study the reaction dynamics of these support materials. The ReaxFF method – which allows for relatively large (>> 10,000 atoms), long-time (>> 1 nanoseconds) fully reactive, fully dynamics simulations of complex materials and their interfaces allows for atomistic-scale simulations that can directly connect to experimental synthesis efforts. In order to expand the ReaxFF capability for support material simulations, we have extended a previously developed ReaxFF parameter set for BaTiO3 to SrTiO3 (STO) – a highly relevant support material for 2D-chalcogenide growth. This ReaxFF description can be used for any Ba/Sr ratio and can describe key surface chemistry – including water vapor and oxidation/reduction reactions at the STO surface. These ReaxFF parameters were validated against Density Functional Theory (DFT) data and was validated be studying ferroelectric transitions in mixed Ba/SrTiO3 materials.
Published in Physical Chemistry Chemical Physics (2021) 23, 25056-25062.
2DCC Role: This project is associated with the 2DCC target of developing new computational capabilities for simulating 2D-materials and support materials associated with the growth of these materials.
What Has Been Achieved: We developed and validated a ReaxFF description for Ba/SrTiO3 perovskites.
Importance of the Achievement: These ReaxFF parameters can be straightforwardly combined with existing ReaxFF descriptions for Mo/W/Se/S/C/O/H, enabling simulations of MoWSeS-material growth on STO support.
Unique Feature(s) of the MIP that Enabled this Achievement: This work responds to a continuing external user interest in ReaxFF parameters for 2D materials and their growth substrates
(If Applicable) Publication: Akbarian, D., Nayir, N. and van Duin, A.C.T. (2021) Understanding the physical chemistry properties of BaxSr1-xTiO3 using ReaxFF based Molecular Dynamics Simulations. Physical Chemistry Chemical Physics 23, 25056-25062.
Acknowledgments: Support for this ReaxFF development at Penn State was provided by the National Science Foundation through the Penn State 2D Crystal Consortium-Materials Innovation Platform (2DCC-MIP) under NSF Cooperative Agreement DMR-1539916 and through AFOSR/MURI grant FA 9550-19-1-0008